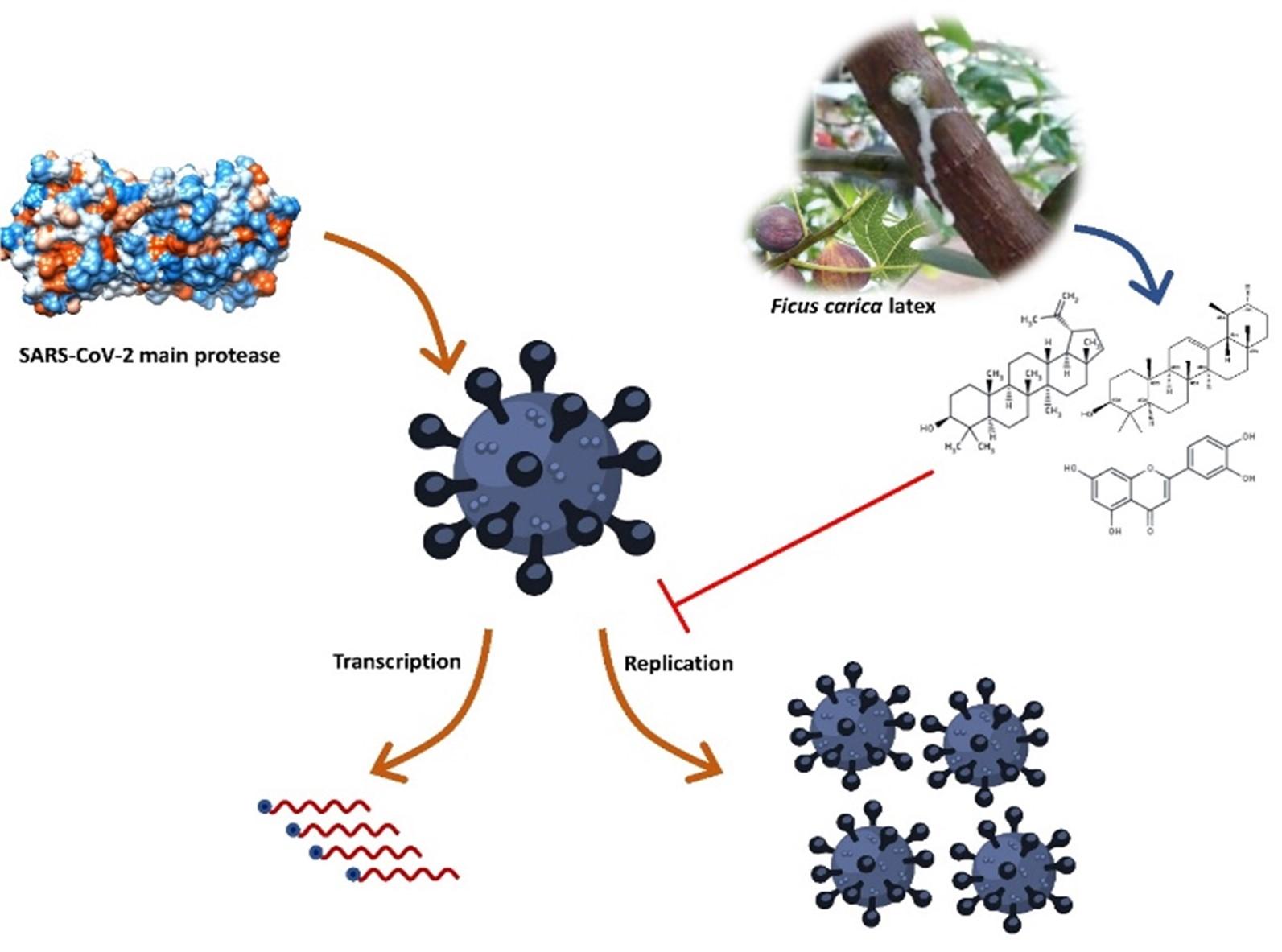
Identification of potential SARS-CoV-2 main protease inhibitors from Ficus Carica Latex: An in-silico approach
Abstract
SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) is the aetiological agent behind the current pandemic of coronavirus disease 2019 (COVID-19). SARS-CoV-2 main protease plays a dynamic role in mediating viral replication and transcription, which is one of the most probable drug targets against SARS-CoV-2. Ficus carica latex encompasses notable bioactive molecules with various biological properties, including antiviral activities. In this study, latex compounds of Ficus carica were screened to find out active phytochemicals against SARS-CoV-2 main protease through molecular docking, molecular dynamics simulation, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiling. A total of 21 compounds were screened, and the compounds, lupeol, α-amyrin, and luteolin, showed the highest binding affinity and intense interaction with the vital catalytic residue His 41 and Cys 145. The molecular dynamics simulation revealed that the amyrin is the most stable compound with higher binding free energy, suggesting that this compound can compete with the native ligands of the main protease. The ADMET analysis indicated that these phytochemicals have considerable physicochemical, pharmacokinetics, and drug-likeness properties and do not possess any considerable detrimental effects and can be considered as potential drug candidates against SARS-CoV-2. However, further in-vitro, in-vivo, and clinical trials are required to observe the exact efficiency of these compounds.
INTRODUCTION
SARS-CoV-2 is like an unexpected nightmare of 2020 that hampered the normal activities of humans and changed the way of life on earth. SARS-CoV-2 is a single-stranded, positive-sense, enveloped virus with a large RNA genome about ~30kb [1, 2]. Unfortunately, there are no specific vaccines or drugs available to treat SARS-CoV-2, although various clinical trials are started to test the feasibility of some drugs. Meanwhile, several existing drugs are repurposed like remdesivir, chloroquine, hydroxychloroquine, lopinavir, and ritonavir against SARS-CoV-2 infection [3, 4]. However, they have primarily proven effective, but still, their efficacy and safety are questionable [5-7]. For these limitations, it is essential to find out the specific drug that would effectively inhibit or reduce SARS-CoV-2 infection.
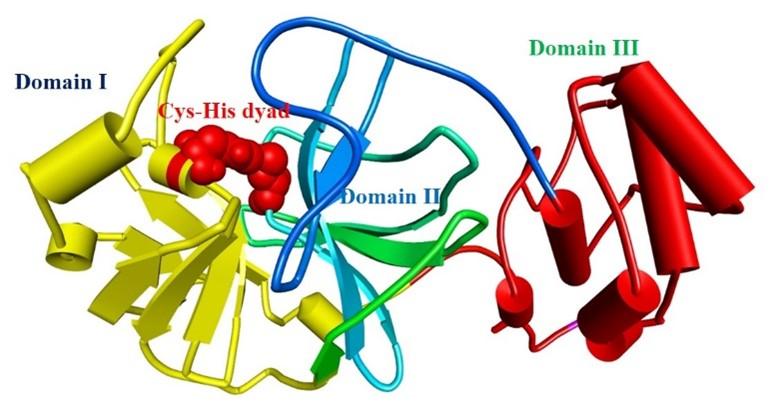
The protease enzyme of SARS-CoV-2 cleaves its native polypeptides and generates active fragments responsible for viral replication, transcription, and translation [8]. These polypeptides are essential for viral replication, transcription, and translation [9, 10]. The main protease (Mpro) catalyzes the SARS-CoV-2 translated polyproteins’ relevance to the viral life cycle and makes them functional [11]. “Mpro has three functional domains; domain I (residues 8–101), domain II (residues 102–184), and domain III (residues 201–303) [12].” “Domain II is joined with domain III by a loop (residues 185–200). The ligand attachment site is situated in the loop of domain I and domain II where catalytic dyad Cys 145-His 41 plays vital roles in ligand management [12]” (shown in figure 1). Mpro also plays a significant role in SARS-CoV-2 replication [12]. Besides, Mpro is not functionally correlated with the human’s homologous proteases, which implies Mpro as a striking target for drug designing [12]. Moreover, several previous studies have shown that Mpro is the prominent target against SARS-CoV-2 infection [13-15]. However, nature provides the best remedy. In previous studies, it has been shown that natural phytochemicals contain active ingredients that might be able to inhibit the SARS-CoV-2 Mpro [14, 16, 17].
The Ficus carica latex (F-latex) encompasses notable molecules with remarkable benefits against antimicrobial-resistant microorganisms along with other biological properties [18, 19]. F-latex contains fatty acids, flavonoids, phenolics, tannins, alkaloids, terpenoids, and sterols with numerous biological benefits [19, 20]. It has been used as traditional medicine around the world and found no cytotoxicity on Vero cells [21]. Several studies found that the F-latex extracts have antibacterial [22, 23], antifungal [22], and antiviral [21] properties. The F-latex compound is active against both DNA and RNA viruses including, adenovirus (ADV), echovirus type 11 (ECV-11) [21], herpes simplex type 1 (HSV-1) [21], HSV-2 [24], caprine herpesvirus-1 (CpHV-1) (showed almost similar efficiency like acyclovir) [25], human papillomavirus (HPV) [26], and the influenza virus H9N2 [27]. As previous studies showed that the F-latex compounds have potential antiviral activity against several viruses along with the same realm, riboviria, so it might be possible to act against SARS-CoV-2 [27]. Therefore, the present study was intended to discover the SARS-CoV-2 Mpro inhibitors from the F-latex compounds to fight against SARS-CoV-2 infection.
MATERIALS AND METHODS
Ficus carica latex compounds profiling and ligand preparation
In this study, we consider Ficus carica latex as a reservoir of SARS-CoV-2 inhibitory compounds. We prepared a dataset of the active phytochemicals of Ficus carica latex from prior studies and the findings. We made a library of bioactive latex compounds by searching related literature in PubMed, google scholar, the web of science, and Scopus databases [28]. We used the PubChem database [29] to download the SDF format of the 3D structures of all compounds and used Open Babel software to convert SDF format compounds to PDB format [30]. We used PyRx [31] integrated mmff94 (Merck molecular force field) force field [32] for optimization and ligand preparation. For further analysis, the ligands were then converted into PDBQT format.
Preparation of receptor
The crystallized structure of Mpro (PDB ID 6LU7 [12]) was curated from the RCSB PDB resource [33]. The water molecules and ligand were removed from the receptor and energy minimized by steepest descent and conjugate gradient techniques. The GROMACS 96 43B1 algorithm in SWISS-PDB viewer [34], and Chimera (Amber Force field) were utilized to prepare the final receptor [35].
Virtual screening
Virtual screening of all compounds was executed through PyRx software by AutoDock wizard [31, 36]. Ligands were considered as flexible, and the protein was considered as rigid during docking. The auto grid engine in AutoDock was used to generate the configuration file of grid parameters (grid box size X, Y, Z; 23, 28, 29, respectively). Potential compounds were considered based on RMSD (root mean square deviation) values. Ligands with most negative docking scores and lower (<1Å) RMSD values were considered for further investigation. The molecular interactions between ligands and receptors were visualized using the Biovia discovery studio visualizer (v 4.5) [37].
Molecular dynamics simulation
In order to get more insights of the protein-ligand complex, molecular dynamics simulation (MD simulation) was performed. The selected complexes were subjected to MD simulation by YASARA molecular dynamics software package using AMBER1 14 forcefield [38, 39] as described formerly [40-42]. The complex was solvated inside the simulation cell using the TIP3 water model [43]. The periodic boundary condition was optimized, and water and Na+/Cl– ions were added. The steepest gradient approach (5000 cycles) was used for each simulation system to minimize energy. We used the PME method to explain the long-range electrostatic interactions within the following conditions 8 Å (electrostatic interaction cut off distance), 298 K temp., 0.9% NaCl, and pH of 7.4 [44]. The simulation time step interval was set to 2 fs [45]. Finally, the simulation was conducted for 20 ns in Berendsen thermostat [46], where the snapshots were saved for every 10 ps. The VMD [47, 48] software was used to calculate RMSD (root mean square deviation), RMSF (root mean square fluctuations), Rg (radius of gyration), and SASA (solvent accessible surface area). Furthermore, the Molecular Mechanics-Poisson–Boltzmann Surface Area (MM-PBSA) binding free energy calculation method was used for all systems to calculate the binding free energy by the following formula,
Binding Energy = EpotRecept + EsolvRecept + EpotLigand + EsolvLigand – EpotComplex – EsolvComplex
ADMET analysis
The compounds that have a higher binding affinity (-12.5 to -7.4) were considered for further ADMET analysis. Based on the canonical SMILES of chosen ligands obtained from PubChem, ADME properties of potent drug candidates were calculated by SwissADME [49], whereas toxicity was analyzed using PreADMET toxicity prediction tools [50].
RESULTS
Virtual screening of retrieved compounds against SARS-CoV-2 main protease
Virtual screening is an effective method to find out probable hits from thousands of compounds [51]. Virtual screening utilizing molecular docking is an alternative way for lead identification in drug discovery [51, 52]. We conducted molecular docking for all (total 21) identified compounds (table 1) with SARS-CoV-2 main protease. The binding affinity of the compounds lies in the following ranges -12.5 to -10.2 kcal/mol, -10.2 to -7.9 kcal/mol, -7.9 to -5.6 kcal/mol, and -5.6 to -5.3 kcal/mol (figure 2). According to the binding affinity, three compounds were selected, namely lupeol (-12.5 kcal/mol), α-amyrin (-7.9 kcal/mol), and luteolin (-7.4 kcal/mol), and considered for further analysis. The a-ketoamide was used as a control ligand in this study as it has been identified recently as a potent inhibitor (binding affinity -7.3 kcal/mol) against SARS-CoV-2 main protease [53]. The α-ketoamide and selected compounds binding affinity, Pubchem Id, formula, IUPAC name, and 2D structure are depicted in table 2.
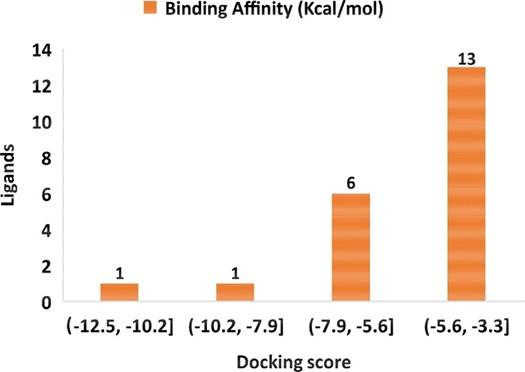
Table 1. Binding affinity of latex compounds of Ficus carica latex.
Table 2. Binding affinity, PubChem Id, formula, binding affinity, IUPAC name, and 2D structure of selected SARS-CoV-2 Mpro inhibitors.
Molecular interaction analysis of selected compounds
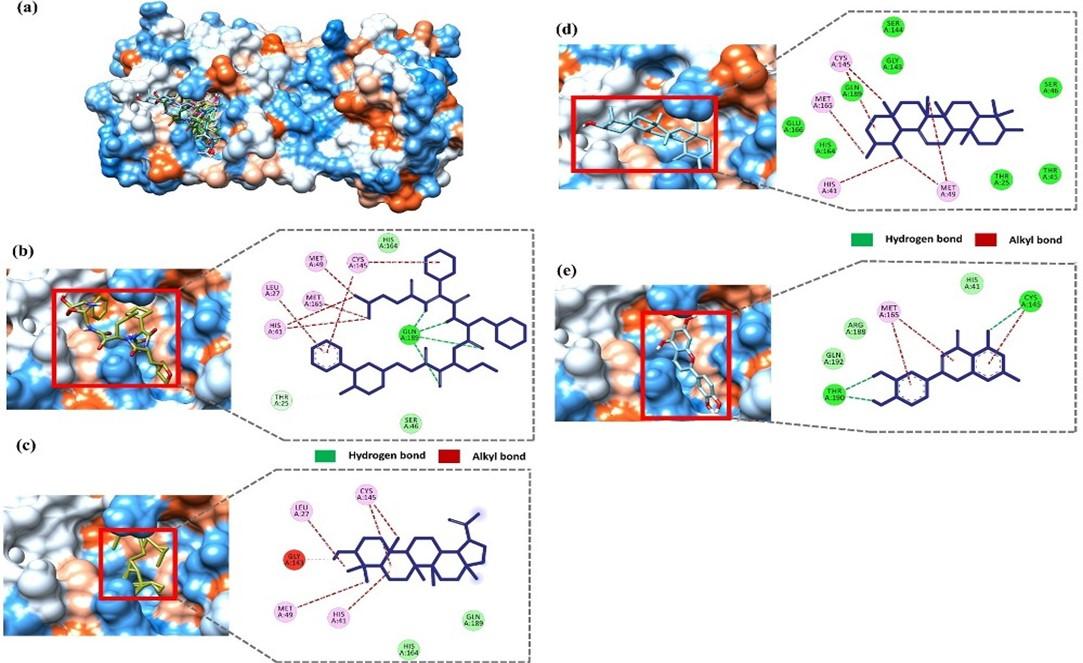
The molecular interactions of the selected compounds were visualized using BIOVIA discovery studio visualizer (v 4.5) [37]. In a recent study, Zhang et al. showed that the inhibitor a-ketoamide forms molecular interactions with His 41, Gly 143, Ser 144, Cys 145, His 163, His 164, Glu 166, Pro 168, and Gln 189 residue of the main protease [53]. Moreover, the native ligand (N3) of our selected Mpro (6LU7) also interacts with the catalytic dyad Cys 145-His 41 [11]. The docked compounds interaction indicates that all three compounds interact with either catalytic residues His 41 and Cys 145 or at least one of them. Moreover, the ligand forms interaction with other substrate-binding pocket residues shown in figure 3. The positive control, a-ketoamide, formed three strong hydrogen bonds with the catalytic loops’ residue Gln 189 and several alkyl bonds with residues Leu 27, His 41, Met 49, Cys145, and Met 165 (figure 3b). Lupeol forms alkyl bonds with Leu 27, His 41, Met 49, and Cys 145 residue (figure 3c). Amyrin forms several alkyl bonds with His 41, Met 49, Cys 145, and Met 165 residue (figure 3d). Luteolin forms hydrogen bonds with Cys 145 and Thr 190 and alkyl bonds with Met 165 residue (figure 3e).
Molecular dynamics simulation
The protein-ligand complex stability was analyzed through MD simulation. We used four parameters to understand complex stability, including RMSD, RMSF, Rg, and SASA. In figure 4a, the positive control α-ketoamide showed the lowest RMSD values compared to all systems, including the apo-proteins. The lower RMSD value illustrates the compactness of the system. The amyrin-protein complex showed a quite similar fashion of stability like apo-protein with stable conformation at 3 ns to 8 ns, and 12 ns to 20 ns. The lupeol also showed stable conformation at 2 ns to 11 ns; after that, a significant structural drifting occurs around 11.5 ns later on, again showed stability from 12 ns to the rest of the simulation. The luteolin complex showed stability at 5 ns to 10 ns, and 18 ns to 20 ns, whereas the rest of the time, it showed more fluctuation. The average RMSD values were 1.43 Å, 1.28 Å, 1.51 Å, 1.66 Å, and 1.98 Å for apo-protein, α-ketoamide-Mpro, amyrin-Mpro, lupeol- Mpro, and luteolin-Mpro complex, respectively (shown in table 3).
In addition, the RMSF calculation was used to analyze the local residual change (shown in figure 4b). The higher RMSF values illustrate the higher flexibility of the system. The α-ketoamide-Mpro complex showed a lower average RMSF value (1.14 Å) compared to all systems. The amyrin- Mpro average RMSF value (1.21 Å) was quite similar to apo-protein RMSF value 1.22 Å. However, lupeol-Mpro and luteolin-Mpro showed 1.35 Å and 1.28 Å RMSF values, respectively (shown in table 3). Besides, the structural compactness was analyzed by calculating the radius of gyration (figure 4c). The lower RG values represent the compactness of the system. Here, the positive control α-ketoamide-Mpro complex showed lower Rg value (22.37 Å) compared to the apo-protein (23.35 Å), while, the amyrin-Mpro complex showed almost same Rg value (22.40 Å) like the positive control. However, the lupeol-Mpro and luteolin-Mpro complex showed lower Rg values, 22.47 Å, and 22.31 Å, respectively, than apo-protein (shown in table 3).
Moreover, the solvent-accessible surface area (SASA) of all systems were analyzed and depicted in figure 4d. The lower SASA value represents the compactness of the systems. The average SASA value of the amyrin-Mpro complex was lower (14059.42 nm2) than the positive control α-ketoamide-Mpro complex (14171.00 nm2). The SASA values of the lupeol-Mpro and luteolin-Mpro complex were 14248.64 nm2 and 14263.55 nm2, respectively (shown in table 3).
Furthermore, the binding free energy calculation depicted that the amyrin showed higher binding energy (225.896 kJ/mol), whereas the lupeol and luteolin showed 181.434 kJ/mol and 51.063 kJ/mol binding free energy, respectively (shown in table 3).
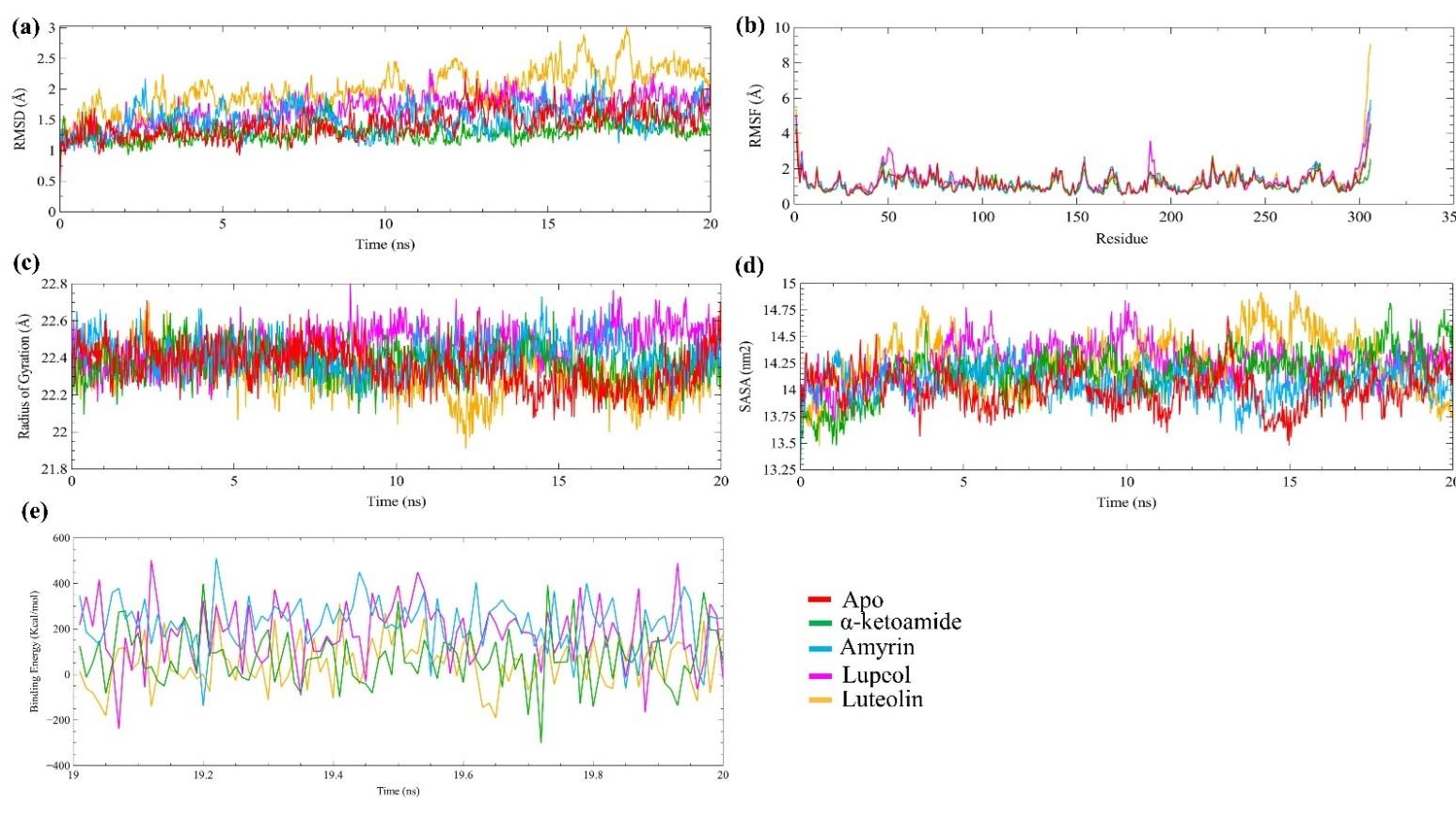
Table 3. The average mean value of MD trajectory.
Table 4. ADMET analysis of top 3 compounds.
ADMET analysis
The different ADMET properties of the selected three compounds are depicted in table 4. In this study, we analyzed physicochemical properties, lipophilicity, water-solubility, pharmacokinetics, drug-likeness, medicinal chemistry, and toxic properties of selected compounds employing ADMET analysis. The pharmacokinetics properties show that the selected compounds cannot permeate the blood-brain barrier (BBB), do not inhibits any cytochrome P450 isoforms (lupeol and α- amyrin), and luteolin shows higher GI absorption, in contrast, lupeol and α-amyrin shows lower GI absorption. In terms of drug-likeness activity, luteolin does not violate Lipinski’s role of five, but both lupeol and α-amyrin violate one role. The toxicity analysis result shows that these compounds do not inhibit the human ether go-go gene (hERG). The hERG gene maintains cardiac systolic and diastolic activity through the potassium ion channel, and inhibition of these channels disrupts the homeostatic balance. The Ames test toxicity data (in TA100 strain-metabolic activation by rat liver homogenate) also shows that these compounds are not mutagenic. However, the detailed ADMET interpretation is explained in the discussion section.
DISCUSSION
Although some drugs are repurposed, there is an urgent need for effective and specific drugs against SARS-CoV-2 infection cause the repurposed drugs have been shown some drastic side effects [54, 55]. Structure-based virtual screening (VS) has been a trend in drug development with all of its underlying computational approaches for more than a decade now, and molecular docking has been thoroughly studied [51]. In this study, we used computer-aided techniques to identify potential inhibitors of SARS-CoV-2 Mpro. The F-latex extracts have been found to inhibit the replication of HSV-1, ECV-11, CpHV-1, and ADV [21, 25]. Moreover., they have been found to act against several drug-resistant pathogens [56].
However, among the 21 studied F-latex compounds, the top three compounds are selected considering their highest binding affinity, and it is found that lupeol, α-amyrin, and luteolin could be used as promising inhibitors against SARS-CoV-2 main protease. According to the binding affinity, lupeol (-12.5 kcal/mol), α-amyrin (-7.9 kcal/mol), and luteolin (-7.4 kcal/mol) have a higher binding affinity than the control α-ketoamide (-7.3 kcal/mol) shown in table 2. Lupeol forms non-covalent alkyl bonds with the crucial catalytic residue His 41 and Cys 145 like α-amyrin. In contrast, luteolin forms one hydrogen and alkyl bonds with Cys 145. Thereby, it may act as inhibitors of SARS-CoV-2 main protease. The selected three compounds form strong non-covalent interactions with other binding site residues. However, similar recent studies also support these findings where the inhibitor compounds form strong covalent and non-covalent bonds with the following residues His 41, Met 49, Tyr 54, Phe 140, His 164, Met 165, Glu 166, Pro168, Asp 187, Arg 188, and Gln189 [57, 58]. Some recent studies suggest that lupeol has antiviral activity against hepatitis B [59], HIV [60], and also reduces pro-inflammatory cytokines [61]. It is also active as an antioxidant, anti-dyslipidemia, anti-hyperglycemic, and anti-mutagenic agent. Besides, it shows hepatoprotective, nephroprotective, neuroprotective, cardioprotective, and anticancer activity in in-vitro and in vivo experiments [62]. Amyrin also has antiviral, anti-inflammatory, and antioxidants properties [59]. The flavonoid luteolin is a potential antiviral agent that is validated by several experiments’ models like as it shows antiviral activity against the tick-borne encephalitis virus model [63], Japanese encephalitis virus [64], HIV-1 [65], and Coxsackie virus B3 [66].
Molecular dynamics simulation acts as a computational microscope to provide bio-molecular insights at progressive and conformational scales [67]. The higher RMSD, RMSF, Rg, and SASA values denote higher flexibility of the system [68, 69]. The α-ketoamide showed lower RMSD, RMSF, and Rg values, which represent its stable binding with SARS-CoV-2 main protease. The higher SASA value of α-ketoamide depicts the system conformation drifting. The amyrin had lesser RMSF, SASA, and Rg values compared to the apo-protein, which depicts its conformational compactness. Besides, the highest binding free energy represents its better binding ability with SARS-CoV-2 main protease. The lupeol showed lower Rg value in contrast with apo-protein and higher binding energy compared to the α-ketoamide, which represents a strong binding, and flexible conformation of this system. Higher RMSD, RMSF, Rg, and SASA values of luteolin represented its higher flexibility compared to all systems. The lupeol and luteolin were the least compacts compounds according to the analysis.
In silico ADMET analysis is a productive, comprehensive, timely, and cost-saving approach to analyze the physicochemical, and drug-likeness properties of any compounds [70]. Computational biology contribution has speeded up drug discovery and is being used to find and develop novel lead compounds against many pathogenic microorganisms and diseases in the biopharmaceutical sector [71, 72]. ADMET analysis provides a clear image of possible drug candidates. The optimal molecular weight of a possible drug should be between 150 to 500 g/mol (Dalton), hydrogen bond acceptor (HBA) ≤ 10, hydrogen bond donor (HBD) ≤ 5, TPSA (topological polar surface area) between 20 and 130 Å2, and molar refractory (MR) range between 40 to 130 [28, 49, 73]. In terms of physicochemical properties, our three selected compounds are in the acceptable range (table 4). The water solubility analysis reveals that luteolin is soluble in water, and the rests two are poorly soluble.
Moreover, these compounds do not penetrate BBB. In new drug discovery approaches, finding out the interaction of drug molecules with cytochrome P450 (CYP) is important cause CYP isoforms maintains the normal cellular metabolism, transformation, and excretion of drugs. Our studied compounds lupeol and α-amyrin do not inhibit any CYP isoforms, which is another good sign of their effectiveness. The medicinal chemistry properties of lupeol and α-amyrin compounds show that they do not possess any PAINS (pan assay interference compounds) alert means they have a high tendency to bind specifically with their targets, and they do not exert any false-positive results. However, lupeol and α-amyrin violate lead likeness properties, but luteolin does not violate lead likeness properties. The studied compounds have moderate synthetic accessibility (value 1 for low and 10 for high synthetic accessibility) [74].
CONCLUSION
In sum, the virtual screening revealed that lupeol, α-amyrin, and luteolin show the highest binding affinity with SARS-CoV-2 main protease and show significant and vital interaction with either both catalytic residues His 41 and Cys 145 or at least one of them. The selected compounds also represent other meaningful interactions like hydrogen bonds, van der Waals bonds, and alkyl bonds. The molecular dynamics simulation study illustrated that among all the phytochemicals, amyrin is the most stable phytochemicals, which could be able to inhibit SARS-CoV-2 main protease strongly. The binding free energy analysis revealed that amyrin and lupeol have higher binding free energy compared to the known SARS-CoV-2 Mpro inhibitor α-ketoamide. The drug-likeness efficiency of these compounds was analyzed by ADMET and reveled that they follow Lipinski’s rule of five and Veber rules with low or no toxicity. Moreover, these compounds cannot penetrate BBB, and in sum, they can be used as drug candidates against SARS-CoV-2.
AUTHORS CONTRIBUTION
RD and MMR conceived the plan of this research. MCA, AJN, MSK wrote the manuscript. MCA analyzed the data and made the figures. RD and MMR, MMK edited the manuscript. All authors revised and approved the manuscript for final submission.
ACKNOWLEDGEMENT
We are thankful to Yeasmin Akter Munni, Graduate Student, Department of Anatomy, Dongguk University College of Medicine, Gyeongju 38066, Republic of Korea, for helping to draw the figures of this project.
CONFLICTS OF INTEREST
The authors declared no competing interests.
References
- [1]Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003;77:8801-11.
- [2]Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Hu Y, et al. Complete genome characterisation of a novel coronavirus associated with severe human respiratory disease in Wuhan, China. bioRxiv. 2020.
- [3]Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269-71.
- [4]Colson P, Rolain JM, Lagier JC, Brouqui P, Raoult D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int J Antimicrob Agents. 2020;55:105932.
- [5]Magagnoli J, Narendran S, Pereira F, Cummings T, Hardin JW, Sutton SS, et al. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. MedRxiv. 2020.
- [6]McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020.
- [7]Mehra MR, Desai SS, Ruschitzka F, Patel AN. RETRACTED: Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis. Lancet (London, England). 2020.
- [8]Ul Qamar MT, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020.
- [9]Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82:2515-27.
- [10]Wang F, Chen C, Tan W, Yang K, Yang H. Structure of Main Protease from Human Coronavirus NL63: Insights for Wide Spectrum Anti-Coronavirus Drug Design. Sci Rep. 2016;6:22677.
- [11]Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289-93.
- [12]Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J Med Chem. 2016;59:6595-628.
- [13]Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2020:1-11.
- [14]Gentile D, Patamia V, Scala A, Sciortino MT, Piperno A, Rescifina A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar Drugs. 2020;18.
- [15]Bacha U, Barrila J, Velazquez-Campoy A, Leavitt SA, Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906-12.
- [16]Cheng B, Li T. Discovery of alliin as a putative inhibitor of the main protease of SARS-CoV-2 by molecular docking. BioTechniques. 2020.
- [17]Ton AT, Gentile F, Hsing M, Ban F, Cherkasov A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol Inform. 2020.
- [18]Ripert C, Pierre A, Jean-Marie B, Anne-Pauline B, Claude G, Mycologie médicale: Tec & doc-Lavoisier; 2013.
- [19]Barolo MI, Ruiz Mostacero N, López SN. Ficus carica L. (Moraceae): an ancient source of food and health. Food Chem. 2014;164:119-27.
- [20]Al-Snafi AE. Nutritional and pharmacological importance of Ficus carica-A. J Pharm. 2017;7:22-48.
- [21]Lazreg Aref H, Gaaliche B, Fekih A, Mars M, Aouni M, Pierre Chaumon J, et al. In vitro cytotoxic and antiviral activities of Ficus carica latex extracts. Nat Prod Res. 2011;25:310-9.
- [22]Rashid KI, Mahdi NM, Alwan MA, Khalid LB. Antimicrobial activity of fig (Ficus carica Linn.) leaf extract as compared with latex extract against selected bacteria and fungi. J Univ Babylon. 2014;22:1620-6.
- [23]AL-Sabawi NA. The Antibacterial Effect of Fig (Leaves Extract and Latex) on Enterococcus faeca-lis as Intracanal Medicament.(An in vi-tro study). Al-Raf Den J. 2010;10:62-71.
- [24]Ay E, Duran N. Investigation of the antiviral activity of Ficus carica L. latex against HSV-2. International Conference on Advanced Materials and Systems (ICAMS): The National Research & Development Institute for Textiles and Leather-INCDTP; 2018. p. 33-7.
- [25]Camero M, Marinaro M, Lovero A, Elia G, Losurdo M, Buonavoglia C, et al. In vitro antiviral activity of Ficus carica latex against caprine herpesvirus-1. Nat Prod Res. 2014;28:2031-5.
- [26]Ghanbari A, Le Gresley A, Naughton D, Kuhnert N, Sirbu D, Ashrafi GH. Biological activities of Ficus carica latex for potential therapeutics in Human Papillomavirus (HPV) related cervical cancers. Sci Rep. 2019;9:1013.
- [27]Asl Najjari AH, Rajabi Z, Vasfi Marandi M, Dehghan G. The effect of the hexanic extracts of fig (Ficus carica) and olive (Olea europaea) fruit and nanoparticles of selenium on the immunogenicity of the inactivated avian influenza virus subtype H9N2. Vet Res Forum. 2015;6:227-31.
- [28]Ali MC, Munni YA, Das R, Sultana M, Akter N, Rahman M, et al. In silico chemical profiling and identification of neuromodulators from Curcuma amada targeting Acetylcholinesterase. bioRxiv. 2020.
- [29]Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47:D1102-d9.
- [30]O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminformatics. 2011;3:33.
- [31]Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243-50.
- [32]Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490-519.
- [33]Burley SK, Berman HM, Bhikadiya C, Bi C, Chen L, Di Costanzo L, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019;47:D464-d74.
- [34]Guex N, Peitsch MC. SWISS‐MODEL and the Swiss‐Pdb Viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714-23.
- [35]Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605-12.
- [36]Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: a review. Biophys Rev. 2017;9:91-102.
- [37]Biovia DS. Discovery studio visualizer v4.5. Release; 2015.
- [38]Dickson CJ, Madej BD, Skjevik ÅA, Betz RM, Teigen K, Gould IR, et al. Lipid14: the amber lipid force field. Dickson CJ, Madej BD, Skjevik AA, et al. Lipid14: The Amber Lipid Force Field. J Chem Theory Comput. 2014;10(2):865-879. doi:10.10212014;10:865-79.
- [39]Krieger E, Vriend G, Spronk CJYo. YASARA–Yet Another Scientific Artificial Reality Application. 2013;993.
- [40]Dash R, Junaid M, Mitra S, Arifuzzaman M, Hosen SMZ. Structure-based identification of potent VEGFR-2 inhibitors from in vivo metabolites of a herbal ingredient. J Mol Model. 2019;25:98.
- [41]Mitra S, Dash R. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP bromodomain inhibitors. J Mol Graph Model. 2018;83:42-52.
- [42]Dash R, Das R, Junaid M, Akash MF, Islam A, Hosen SZ. In silico-based vaccine design against Ebola virus glycoprotein. Adv appl bioinformatics chem. 2017;10:11-28.
- [43]Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein MLJTJocp. Comparison of simple potential functions for simulating liquid water. J Chem Phy. 1983;79:926-35.
- [44]Krieger E, Nielsen JE, Spronk CA, Vriend GJJomg, modelling. Fast empirical pKa prediction by Ewald summation. J Mol Graph Model. 2006;25:481-6.
- [45]Krieger E, Vriend GJJocc. New ways to boost molecular dynamics simulations.J Comput Chem. 2015;36:996-1007.
- [46]Berendsen HJ, Postma Jv, van Gunsteren WF, DiNola A, Haak JRJTJocp. Molecular dynamics with coupling to an external bath. J Chem Phy. 1984;81:3684-90.
- [47]Humphrey W, Dalke A, Schulten KJJomg. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33-8.
- [48]Dash R, Das R, Junaid M, Akash MFC, Islam A, Hosen SZJA, et al. In silico-based vaccine design against Ebola virus glycoprotein. Adv Appl Bioinform Chem. 2017;10:11.
- [49]Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
- [50]Lee S, Lee I, Kim H, Chang G, Chung J, No K. The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. EuroQSAR. 2003.pp. 418-420.
- [51]Kontoyianni M. Docking and Virtual Screening in Drug Discovery. Methods Mol Biol. 2017;1647:255-66.
- [52]Neves BJ, Braga RC, Melo-Filho CC, Moreira-Filho JT, Muratov EN, Andrade CH. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. FFront Pharmacol. 2018;9:1275.
- [53]Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269-71.
- [54]Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet (London, England). 2020;395:1569-78.
- [55]Enmozhi SK, Raja K, Sebastine I, Joseph J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: an in silico approach. J Biomol Struct Dyn. 2020:1-7.
- [56]Aref HL, Salah K, Chaumont JP, Fekih A, Aouni M, Said K. In vitro antimicrobial activity of four Ficus carica latex fractions against resistant human pathogens (antimicrobial activity of Ficus carica latex). Pak J Pharm Sci. 2010;23:53-8.
- [57]Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331-5.
- [58]Jin Z, Zhao Y, Sun Y, Zhang B, Wang H, Wu Y, et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol. 2020;27:529-32.
- [59]Parvez MK, Alam P, Arbab AH, Al-Dosari MS, Alhowiriny TA, Alqasoumi SI. Analysis of antioxidative and antiviral biomarkers β-amyrin, β-sitosterol, lupeol, ursolic acid in Guiera senegalensis leaves extract by validated HPTLC methods. Saudi Pharm J. 2018;26:685-93.
- [60]Esposito F, Mandrone M, Del Vecchio C, Carli I, Distinto S, Corona A, et al. Multi-target activity of Hemidesmus indicus decoction against innovative HIV-1 drug targets and characterization of Lupeol mode of action. Pathog Dis. 2017;75.
- [61]Ahmad SF, Pandey A, Kour K, Bani S. Downregulation of pro-inflammatory cytokines by lupeol measured using cytometric bead array immunoassay. Phytother Res. 2010;24:9-13.
- [62]Tsai FS, Lin LW, Wu CR. Lupeol and Its Role in Chronic Diseases. Adv Exp Med Biol. 2016;929:145-75.
- [63]Krylova NV, Popov AM, Leonova GN, Artiukov AA, Maĭstrovskaia OS. [Comparative study of antiviral activity of luteolin and 7,3′-disulfate luteolin]. Antibiot Khimioter. 2011;56:7-10.
- [64]Fan W, Qian S, Qian P, Li X. Antiviral activity of luteolin against Japanese encephalitis virus. Virus Res. 2016;220:112-6.
- [65]Mehla R, Bivalkar-Mehla S, Chauhan A. A flavonoid, luteolin, cripples HIV-1 by abrogation of tat function. PloS one. 2011;6:e27915.
- [66]Wu S, Wang HQ, Guo TT, Li YH. Luteolin inhibits CVB3 replication through inhibiting inflammation. J Asian Nat Prod Res. 2019:1-12.
- [67]Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE. Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys. 2012;41:429-52.
- [68]Dash R, Choi HJ, Moon IS. Mechanistic insights into the deleterious roles of Nasu-Hakola disease associated TREM2 variants. Sci Rep. 2020;10:1-14.
- [69]Dash R, Ali MC, Dash N, Azad MAK, Hosen SMZ, Hannan MA, et al. Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding. Int J Mol Sci. 2019;20.
- [70]Talele TT, Khedkar SA, Rigby AC. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem. 2010;10:127-41.
- [71]Hirono S. [An introduction to the computer-aided structure-based drug design–applications of bioinformatics to drug discovery]. Jpn J Clin Pathol. 2002;50:45-51.
- [72]Ivanov AS, Veselovsky AV, Dubanov AV, Skvortsov VS. Bioinformatics platform development: from gene to lead compound. Methods Mol Biol. 2006;316:389-431.
- [73]Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv. 1997;23:3-25.
- [74]Rahman A, Naheed NH, Raka SC, Qais N, Momen AZMR. Ligand-based virtual screening, consensus molecular docking, multi-target analysis and comprehensive ADMET profiling and MD stimulation to find out noteworthy tyrosine kinase inhibitor with better efficacy and accuracy. Orient Pharm Exp Med. 2019. doi.org/10.1007/s13596-019-00406-9.